

Introduction
The EU MDR is a new set of regulations that came into effect on 26th May 2021, following a 4-year transition period after being adopted in May 2017. It aims to improve the safety and performance of the medical devices throughout their lifecycle by tightening up the requirements and adding more emphasis on the market vigilance which was missing when the previous MDD (Medical Devices Directive) law was passed in 1992.
Similarly on 26th May 2022, the in vitro Diagnostic Medical Devices Regulation (EU IVDR) will come into effect for all the EU member states. However, the EU MDR & EU IVDR will not apply in Great Britain. The UK’s regulation for medical devices will continue to act based on MDR 2002 for now, which was implemented in the UK by the Medical Devices Regulation 2002.
Key Upcoming Dates:
26th May 2024 - End of MDR transitional period
26th May 2025 - All IVDD certificates to become void
26th May 2027 - End of IVDR transitional period
The need to setup a new MDR
When the MDD came into force in 1992, there was no concept of software as a medical device (SaMD) at that time. Other technology advancements throughout the supply chain have also enhanced general traceability and transparency as well.
Also, an aging population in Europe needed the devices to not only pass clearance to be sold in the market but to also provide enhanced usability and transparent technical information for increased patient safety.
Although the renewal of MDD certification is still allowed until 26th May 2024, devices would ultimately need to be compliant with the MDR. This increased time is to allow for a smooth transition without market disruption.
Understanding the grace period
A second set of corrections was published by the Council of the European Union for the MDR which gives manufacturers of certain Class I low risk devices under the MDD to be compliant until the 25th of May 2024, provided that these devices do not have any significant changes.
This additional time can benefit the manufacturers of Class I devices by allowing them time to make the necessary revisions and complete an MDD CE certification. Which means that all devices approved and lawfully placed on the market can continue to be sold until 26th May 2024.
Industry readiness
Supply chain traceability technologies such as serialization can and must play a key role in overseeing, improving and assuring the quality of medical devices, just as it does for pharmaceutical products. By tracking individual devices throughout their life cycle, traceability makes it possible for all stakeholders at every step along the supply chain - from the manufacturer to the healthcare provider to the patient, in order to know everything there is to know, about any given device, anywhere in the world.
Many manufacturers are expressing interest in ‘connecting’ their devices and taking advantage of artificial intelligence (AI) and other available technologies. That being said, both the medical device industry and its regulatory bodies have yet to fully embrace serialization, perhaps because the conversation is still in the exploratory stage.
EUDAMED
European database on medical devices (EUDAMED) is an IT system developed by the European Commission to bring into force the requirements of MDR 2017/745 and IVDR 2017/746. EUDAMED aims to improve the coordination of information on medical devices in the EU member states by enhancing overall transparency and including better access to information for the public and healthcare professionals.
The new EUDAMED database replaces the existing database of eudamed2 which was implemented under the previous medical devices regulation. The new database is expected to be much larger as compared to the previous one and will be used for storing information on the devices, registration of manufacturers, authorized representatives, information regarding certificates and clinical investigations.
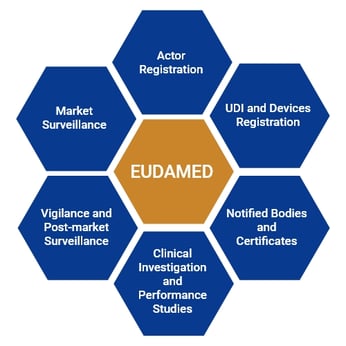
EUDAMED Modules
EUDAMED consists of 6 modules which are interrelated to each other. These are:
- Actor registration
- UDI and Devices registration
- Notified Bodies and Certificates
- Clinical Investigation and performance studies
- Vigilance and post-market Surveillance
- Market Surveillance
Out of these 6 modules, the Actor registration module is available since December 2020, while the module on UDI and device registration (second module) and the module on Notified Bodies and Certificates (third module) are available since October 2021. The remaining modules along with the mechanism for scrutiny and the clinical evaluation consultation procedure (CECP) is expected to release when EUDAMED is fully functional.
- Actor Registration Module:
EUDAMED has defined different Actor roles where an actor is a legal person or organization with a specific role that has to be registered with the EUDAMED. The roles are divided into two parts, the supervising entities & the economic operators. The supervising entities consists of:
- Commission (EC)
- Competent Authority (CA)
- Designating Authority (DA)
- Notify Body (NB)
While the economic operators consist of:
- Manufacturer (MF)
- Authorised Representative (AR)
- System & Procedure Pack Producer (PR)
- Importer (IM)
Each economic operators receive an Actor ID/SRN (Single Registration Number) once the relevant competent authority has validated the Actor registration request. This Actor ID/SRN uniquely identifies the economic operators in EUDAMED. It consists of the Country ISO2 Code, the Actor Role Abbreviation, and a nine-digit number (BE-MF/AR/PR/IM-Number). In case the economic operators have multiple roles, they need to ensure that separate registration requests are submitted to obtain a specific Actor ID/SRN for each role.
For non-EU manufacturers, their authorized representative performs this process on their behalf.
- UDI and Devices registration
This module is used to upload the UDI/Device information of all devices/products that the manufacturer of medical devices wants to place on the market. This allows easier traceability, improved safety, and better monitoring of the medical devices by the authorities. The following information needs to be uploaded as a part of this module:
- Basic UDI-DI (This is the main access key for device-related information in the EUDAMED database)
- UDI-DI (This acts as the main identifier of a medical device as it identifies the specific device within a given product family)
- Package UDI-DI (This is not always applicable, it is assigned to the higher package as an additional level UDI-DI)
- Notified Bodies and Certificates
Until EUDAMED is fully functional, this module will be used by the notified bodies to register certificates and Summaries of Safety and Clinical Performance (SSCP) only on voluntary basis if all the parties referenced in the certificates are already registered, also on a voluntary basis. The objective of this module is to enable communication between the notified bodies and make all the required information accessible to public.
The Way Forward
Even if your company is compliant with MDD, MDR brings in many new regulations and replaces many others, so it is important to make sure the devices are compliant way before the deadline. The new regulation also states the obligation of manufacturers and distributors to have a stricter clinical evaluation & conformity assessment procedure in place while focusing on the risk management and quality management systems at the same time.
EUDAMED database is also slowly developing with 3 modules released till date. It is advised to go through the functional specification documents and other documents available on EUDAMED’s website (Overview | Public Health (europa.eu)) for these modules to prepare yourselves for the new regulation and start the transition from MDD to the MDR soon enough as the last minute adjustments could affect the business continuity for devices that have a large inflow into the market. Medical-device players, facing unique opportunities and headwinds, will need to reinvent themselves as digital health companies to remain relevant and win in this fast-evolving market.
How can CosmoTrace help?
We provide serialization consulting, implementation & integration services to help our clients with managing the end-to-end serialization projects and preparing them for the existing and upcoming regulations across the globe.
We are well versed with the Medical Devices regulations and recent standards for various markets and can help you with implementing the project to meet the upcoming deadlines.
Our team of experts strategize and plan end-to-end solutions using a combination of years of knowledge in product serialization, pharmaceutical supply chains, life sciences and brand integrity.
Disclaimer
This information is being provided ‘As Is’ with no claims of suitability for a particular purpose. It represents just one possible interpretation of information available in the public domain or through membership organizations, and that interpretation is subject to change. This information does not constitute legal advice. Users must refer to the source material for the complete requirements and form their own interpretation before making business decisions. Please use the references below to follow the updates at the source.

Reach out to CosmoTrace for support getting your technical documents ready for EUDAMED registration, MDD renewal or the MDD to MDR transition. We have expert medical writers to support your organization with the implementation of regulation, project management, risk management, quality management, consulting, and reporting to the authorities.